TRANSPORTE DE FLUIDOS Y ELECTROLITOS EN EL INTESTINO: BASES FISIOLÓGICAS Y ENFERMEDADES ASOCIADAS
TRANSPORT OF FLUIDS AND ELECTROLYTES IN THE INTESTINE: PHYSIOLOGICAL BASIS AND ASSOCIATED DISEASES
Autores: César Hoyos Cabrera1; Luis Iraita Cavero1; José Javier Alcantara1; Jesús Medina Mendonza1; Pedro Medina Panta1
1. Universidad Nacional del Santa, Nuevo Chimbote, Perú
Correspondencia:
José Javier Alcantarpa
Correo: 202224047@uns.edu.pe
ORCID
César Hoyos C 0009-0007-3919-7372
Luis Iraita C 0009-0008-8549-4586
José Javier A 0009-0009-3388-8177
Jesús Medina M 0009-0004-4612-0849
Pedro Medina P 0009-0009-4944-7288
Declaración de autoría
Todos los autores declaran haber participado en el desarrollo y diseño del estudio.
Declaración de conflicto de interés
Los autores declaran no tener conflictos de interés relacionados con el estudio.
Declaración de financiamiento
El presente estudio no recibió ningún tipo de
financiamiento externo o interno para su desarrollo
RESUMEN:
Describir los mecanismos fisiológicos del transporte intestinal de fluidos y electrolitos, su regulación y su implicancia en enfermedades digestivas. La revisión narrativa de literatura fisiológica y biomédica reciente sobre los mecanismos de absorción y secreción intestinal de agua y electrolitos, enfocándose en los transportadores y canales principales (SGLT1, NHE3, DRA, ENaC, CFTR) y su control neuroendocrino. El transporte intestinal depende de la interacción entre mecanismos activos y pasivos regulados por señales neurohormonales. Alteraciones en estos sistemas causan trastornos como la diarrea secretora, la enfermedad de Crohn y el síndrome del intestino corto. En la diarrea secretora, las enterotoxinas activan CFTR y aumentan la secreción de Cl⁻ y agua; en Crohn, la inflamación altera la absorción y la integridad epitelial; y en el intestino corto, la resección induce adaptaciones funcionales y estructurales compensatorias. Comprender los fundamentos del transporte intestinal permite explicar la fisiopatología de diversas enfermedades y desarrollar estrategias terapéuticas efectivas como la rehidratación oral, la nutrición enteral precoz y el uso de moduladores hormonales.
Palabras clave: transporte intestinal; electrolitos; diarrea secretora; enfermedad de Crohn; síndrome de intestino corto.
ABSTRACT:
To describe the physiological mechanisms of intestinal fluid and electrolyte transport, their regulation, and implications in digestive diseases. A narrative review of recent physiological and biomedical literature was conducted, focusing on major intestinal transporters and channels (SGLT1, NHE3, DRA, ENaC, CFTR) and their neuroendocrine regulation. Intestinal transport depends on the interplay between active and passive mechanisms regulated by neurohormonal signals. Dysregulation causes disorders such as secretory diarrhea, Crohn’s disease, and short bowel syndrome. In secretory diarrhea, enterotoxins activate CFTR, increasing Cl⁻ and water secretion; in Crohn’s disease, inflammation impairs absorption and barrier integrity; and in short bowel syndrome, resection induces compensatory functional and structural adaptations. Understanding intestinal transport physiology clarifies the pathophysiology of major gastrointestinal diseases and guides effective therapeutic strategies such as oral rehydration, early enteral nutrition, and hormonal modulators.
Keywords: intestinal transport; electrolytes; secretory diarrea; Crohn’s
disease; short bowel syndrome.
INTRODUCCIÓN
El transporte de fluidos y electrolitos en el intestino es un proceso de gran importancia para la vida, ya que garantiza la absorción adecuada de agua, sales minerales y nutrientes, manteniendo el equilibrio hidroelectrolítico y la homeostasis del organismo. Aunque la ingesta diaria de agua en la dieta es limitada, el aparato digestivo maneja volúmenes mucho mayores gracias a las secreciones gastrointestinales, que suman entre 8 y 10 litros al día. La eficiente reabsorción de este volumen depende de una interacción precisa entre mecanismos de transporte activos y pasivos, regulados por factores hormonales, nerviosos y paracrinos, y llevados a cabo por estructuras especializadas de la mucosa intestinal.
La comprensión de estos procesos ha avanzado considerablemente en las últimas décadas, permitiendo identificar transportadores y canales clave, como SGLT1, NHE3, DRA, ENaC y CFTR, cuya actividad coordinada asegura la absorción y secreción equilibrada de agua y electrolitos. Sin embargo, la alteración de estos mecanismos, ya sea por infecciones, enfermedades inflamatorias, mutaciones genéticas o resecciones quirúrgicas extensas, puede conducir a graves trastornos clínicos como diarrea secretora, enfermedad inflamatoria intestinal, y síndrome de intestino corto.
En este contexto, el presente artículo revisa los fundamentos
fisiológicos del transporte intestinal de fluidos y electrolitos, describe los
principales mecanismos celulares y moleculares implicados, y analiza cómo su
disfunción contribuye a la fisiopatología de diversas enfermedades, resaltando
la relevancia de estos conocimientos para el diagnóstico, tratamiento y
prevención de dichas afecciones.
MARCO TEÓRICO
El intestino cumple una función de suma importancia en la absorción de agua, electrolitos y nutrientes, procesos sustanciales para mantener la homeostasis del organismo. A pesar de que la ingesta oral diaria de agua es de 1,5 a 2,5 litros, las secreciones digestivas elevan la carga hídrica intestinal a unos 8,5 litros, de los cuales el intestino delgado absorbe la mayor parte y el colon reabsorbe casi todo el resto, dejando menos de 200 mL en las heces. Este equilibrio se logra mediante mecanismos de transporte pasivo y activo, regulados por factores hormonales, nerviosos y paracrinos, y gracias a la organización estructural de criptas y vellosidades. Cualquier alteración en estos procesos puede provocar diarrea, deshidratación o desequilibrio electrolítico con consecuencias graves (1).
1. Mecanismos fisiológicos del transporte intestinal
La mucosa intestinal está formada por una diversidad de tipos celulares que cumplen funciones especializadas. Los enterocitos constituyen la principal célula absortiva y cubren la mayor parte de la superficie vellosa, siendo responsables de la absorción activa y pasiva de nutrientes, agua y electrolitos. Las células caliciformes secretan moco, esencial para la protección y lubricación de la superficie epitelial, mientras que las células de Paneth participan en la defensa antimicrobiana mediante la secreción de péptidos antimicrobianos. Otras células especializadas, como las células enteroendocrinas, liberan hormonas que regulan la motilidad y secreción intestinal (1).
La función de transporte intestinal depende de la polarización de la membrana plasmática de los enterocitos: la membrana apical (en contacto con la luz intestinal) alberga transportadores encargados de la absorción de nutrientes y iones, mientras que la membrana basolateral (en contacto con el intersticio y vasos sanguíneos) contiene bombas y canales que mantienen los gradientes electroquímicos y permiten la salida de los productos absorbidos hacia la circulación sistémica. Entre los mecanismos de transporte, destacan el transporte pasivo (difusión simple y facilitada, ósmosis) y el transporte activo (primario y secundario), fundamentales para la absorción y secreción de solutos (2).
En la base de estos procesos está la bomba Na⁺/K⁺ ATPasa, localizada en la membrana basolateral, que mantiene el gradiente de sodio necesario para la absorción activa secundaria de nutrientes e iones, extruyendo tres Na⁺ hacia el intersticio e introduciendo dos K⁺ al citosol a expensas de ATP (1). Los cotransportadores Na⁺/glucosa (SGLT1) y Na⁺/aminoácidos, presentes en la membrana apical, utilizan este gradiente para facilitar la entrada simultánea de sodio y nutrientes desde la luz intestinal. El intercambiador Cl⁻/HCO₃⁻ contribuye a la absorción electroneutra de NaCl y a la secreción de bicarbonato, clave para el balance ácido-base (2) (Figura 1).
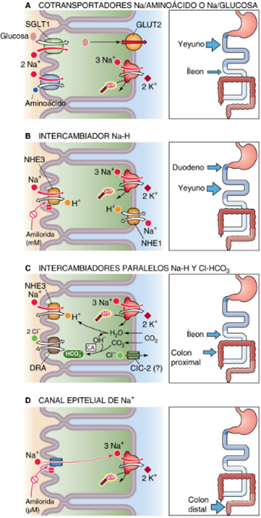
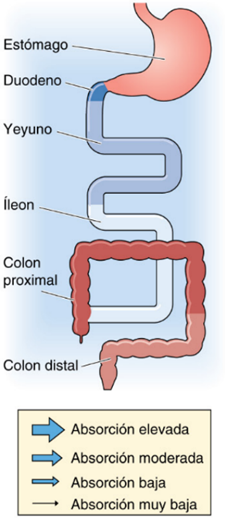
Figura 1. Resumen de los mecanismos de transporte principales para Na⁺, Cl⁻, HCO₃⁻ y agua en las células epiteliales intestinales.
En cuanto a canales, el CFTR, regulado por AMPc, es el principal canal de Cl⁻ en la membrana apical, implicado en la secreción de cloruro y agua, y su disfunción causa patologías como la fibrosis quística. El ENaC (epithelial sodium channel) también participa en la absorción electrogénica de Na⁺, especialmente en el colon distal (3).
La regulación del transporte intestinal es compleja e involucra señales neurohormonales (acetilcolina, VIP, noradrenalina), hormonas locales (secretina, CCK, somatostatina) y mediadores paracrinos como prostaglandinas y óxido nítrico, que modulan la actividad de transportadores y canales a través de segundos mensajeros intracelulares (AMPc, Ca²⁺) (1).
2. Mecanismos de absorción y secreción en intestino delgado y colon
El proceso de absorción y secreción intestinal es esencial para el mantenimiento de la homeostasis corporal y varía considerablemente entre el intestino delgado y el colon debido a diferencias estructurales y funcionales. El intestino delgado es el principal sitio de absorción de agua, electrolitos y nutrientes, caracterizándose por la absorción isoosmótica, es decir, el agua se transporta en proporción directa a la absorción de solutos, especialmente sodio y cloruro. El gradiente osmótico se genera principalmente a través del transporte activo de Na⁺ desde la luz intestinal hacia el interior del enterocito, lo que arrastra agua de forma pasiva, permitiendo una absorción eficiente y ajustada a las necesidades del organismo (1).
3. Mecanismos celulares de absorción de NA+
Las células epiteliales de las vellosidades del intestino delgado y las superficiales del colon absorben la mayor parte del Na⁺ mediante cuatro mecanismos principales que actúan en la membrana apical. En todos los casos, la bomba Na⁺/K⁺-ATPasa, situada en la membrana basolateral, impulsa el paso del Na⁺ hacia la sangre y genera el gradiente electroquímico que facilita su entrada desde la luz intestinal (4).
3.1. Cotransporte Na/glucosa y Na/aminoácido en intestino delgado
La absorción de Na+ tiene lugar únicamente en las células epiteliales de las vellosidades. Este proceso es el principal mecanismo de absorción de Na+ tras una comida. Está mediada por proteínas de transporte específicas de la membrana apical, las cuales captan los nutrientes acoplados a Na+ (a favor del gradiente de Na+ y en contra del gradiente de los nutrientes), mediante transporte activo secundario, aumentando la [Na+]i (2).
3.2.Intercambio Na/H en duodeno y yeyuno
El HCO3– (producto de las secreciones pancreática, biliar y duodenal) aumenta la absorción de Na+ en la parte proximal del intestino delgado ya que estimula el intercambio Na-H. Este intercambiador acopla la captación de Na+ a través de la membrana apical con la salida de H+ hacia la luz intestinal. La energía requerida para esta acción proviene del gradiente de Na+. El intercambiador Na-H posee varias isoformas diferentes que se encuentran en las membranas apicales (NH2 y NH3) y basolaterales (NH1) (2).
3.3. Intercambio paralelo de Na-H y Cl-HCO3 en íleon y colon proximal
La absorción electroneural de NaCl, que tiene lugar en el íleon y en todo el intestino grueso, es resultado de intercambiadores paralelos Na-H y Cl-HCO3– de la membrana apical. En el colon, el intercambio Cl-HCO3 está mediado por DRA (2).
3.4. NaC en porción distal del colon
La absorción de Na+ tiene lugar a través la membrana apical por medio de canales epiteliales de Na+ (ENaC), con especificidad elevada por este catión. A nivel de colon, esta absorción es muy efectiva y va en contra de su gradiente de concentración (2).
4. Mecanismos celulares de absorción y secreción de CL−
El transporte de cloruro (Cl⁻) ocurre tanto en el intestino delgado como en el grueso, y puede estar acoplado al sodio (Na⁺) o regulado de forma independiente, siguiendo rutas paracelulares o transcelulares, según el gradiente electroquímico o los mecanismos de intercambio presentes en cada segmento intestinal (4).
4.1. La absorción voltaje-dependiente de Cl⁻
Se genera un potencial eléctrico negativo en la luz intestinal debido a la absorción electrogénica de Na⁺. En el intestino delgado, esta diferencia de potencial se origina principalmente por el transporte de Na⁺ acoplado a glucosa y aminoácidos durante el periodo posprandial, mientras que en el colon distal es inducida por los canales epiteliales de sodio (ENaC). En ambos casos, el Cl⁻ se absorbe pasivamente, principalmente por vía paracelular, sin intervención de mecanismos activos ni sensibilidad a nucleótidos cíclicos o calcio intracelular (2).
4.2.El intercambio electroneutral Cl⁻/HCO₃⁻
Permite la absorción de cloruro y la secreción de bicarbonato en el íleon y el colon. Este mecanismo se realiza a través del intercambiador apical DRA (SLC26A3), que realiza un intercambio 1:1 entre Cl⁻ luminal e HCO₃⁻ intracelular. Aunque no se conoce completamente la vía basolateral de salida del Cl⁻, se sugiere la participación del canal ClC-2. Este proceso ocurre predominantemente en las células de las vellosidades y epiteliales superficiales, sin requerir la participación de intercambios paralelos de Na⁺/H⁺ (2).
4.3. El intercambio paralelo Na-H y Cl-HCO3 en el íleon y en el colon
La absorción electroneutral de NaCl, que se relaciona con el transporte de sodio también participa en la captación de cloruro (Cl⁻) a nivel del íleon y del colon proximal. En este mecanismo, el paso del Cl⁻ a través de la membrana apical ocurre mediante dos intercambiadores que operan de forma paralela: el de sodio-protones (Na⁺/H⁺), NHE3 (o SLC9A3), y el de cloruro-bicarbonato (Cl⁻/HCO₃⁻), DRA (o SLC26A3). Ambos sistemas están funcionalmente acoplados a través de la regulación del pH intracelular (2).
4.5. La secreción electrogénica de Cl⁻
Ocurre principalmente en las criptas del intestino delgado y grueso. Aunque es mínima en condiciones basales, puede incrementarse de forma intensa ante la acción de secretagogos como la acetilcolina, VIP, enterotoxinas bacterianas, histamina o laxantes. Esta secreción se basa en la acción coordinada de una bomba Na⁺/K⁺ ATPasa, un cotransportador basolateral NKCC1 (Na⁺/K⁺/2Cl⁻), y canales de K⁺ que mantienen el gradiente necesario. El Cl⁻ se libera hacia la luz intestinal mediante el canal CFTR, cuya activación depende de rutas intracelulares que elevan AMPc, GMPc o Ca²⁺. Este proceso favorece también la secreción pasiva de Na⁺ y agua por vía paracelular, generando una secreción neta de NaCl y líquidos (2).
Por otro lado, el colon cumple funciones cruciales en la conservación de agua y electrolitos y en la formación de heces. Su capacidad de absorción fina y selectiva permite reabsorber hasta 1,9 litros de agua por día, dejando menos de 200 mL para ser eliminados en las heces. El principal mecanismo de absorción de Na⁺ en el colon distal es el canal de sodio epitelial (ENaC), regulado por la aldosterona, lo que permite una absorción electrogénica y ajustable de acuerdo con las necesidades del organismo (5). Paralelamente, el colon regula la secreción de K⁺ y Cl⁻; el primero se secreta activamente a través de canales específicos bajo influencia de hormonas y del estado ácido-base, mientras que la secreción de Cl⁻ está mediada principalmente por el canal CFTR y juega un papel importante en la hidratación de las heces (6).
Las diferencias fisiológicas entre intestino delgado y colon radican en que el primero está especializado en la absorción masiva e isoosmótica de nutrientes y agua, con una gran superficie y múltiples transportadores apicales, mientras que el colon se especializa en la concentración y almacenamiento de residuos, la reabsorción fina de agua y electrolitos, y la regulación de la excreción de potasio (1).
4.6. Regulación del transporte de fluidos y electrolitos
Los factores que regulan el transporte intestinal pueden dividirse en locales y sistémicos. A nivel local, el contenido luminal incluyendo su osmolaridad, la presencia de ácidos biliares, metabolitos bacterianos y el pH que modula directamente la actividad de transportadores epiteliales. Por su parte, las señales sistémicas relacionadas con el volumen extracelular, la presión arterial y la composición iónica del plasma inducen respuestas hormonales adaptativas (1).
Entre las principales hormonas implicadas en la regulación del transporte intestinal se encuentran la aldosterona, el cortisol y los péptidos natriuréticos. La aldosterona, secretada por la corteza suprarrenal en respuesta a estados de hipovolemia o hiponatremia, incrementa la expresión de ENaC y de la Na⁺/K⁺ ATPasa en el colon, promoviendo la reabsorción de sodio y agua, primordial para preservar el volumen extracelular (8). El cortisol, además de su función en el metabolismo energético, actúa de manera permisiva sobre la acción de otras hormonas y modula directamente la expresión de transportadores como SGLT1, además de influir sobre el tono vascular intestinal. En situaciones de estrés crónico, puede alterar la función de barrera epitelial, afectando indirectamente el transporte hidroelectrolítico (7). Por otro lado, los péptidos natriuréticos, como el ANP y BNP, ejercen un efecto antagónico al de la aldosterona al elevar los niveles de GMPc intracelular, inhibiendo así la actividad de ENaC y promoviendo la eliminación de sodio en situaciones de sobrecarga de volumen (8).
El sistema nervioso entérico (SNE), también conocido como el “segundo cerebro”, juega un papel clave en la regulación autónoma del transporte intestinal. A través de los plexos mientérico y submucoso, controla no solo la motilidad y el flujo sanguíneo, sino también la secreción de fluidos. Neurotransmisores como la acetilcolina (ACh), la serotonina (5-HT) y el péptido vasoactivo intestinal (VIP) modulan la apertura de canales iónicos como el CFTR, facilitando así la secreción de cloro, sodio y agua. En el caso del VIP aumenta los niveles de AMPc en las células epiteliales, activando CFTR y promoviendo una secreción neta de electrolitos. Esta acción es particularmente importante en la fase postprandial, pero puede verse exacerbada en condiciones patológicas como el cólera o el síndrome carcinoide. Además, el SNE mantiene un diálogo bidireccional con el sistema inmunológico, lo que amplifica su papel regulador en enfermedades crónicas como la enfermedad inflamatoria intestinal (9).
En condiciones fisiológicas, el intestino adapta su actividad secretora y absortiva según el estado nutricional. Durante la digestión, predominan mecanismos que favorecen la absorción de agua y sodio, mediados por transportadores como SGLT1 y NHE3. En cambio, durante el ayuno, se activan mecanismos hormonales principalmente mediados por la aldosterona que regulan la reabsorción en el colon distal. En contraste, diversas condiciones patológicas pueden alterar profundamente esta regulación. En el caso de las diarreas secretoras inducidas por enterotoxinas de Vibrio cholerae, se produce una elevación masiva de AMPc o GMPc, que activa el canal CFTR y conlleva una pérdida excesiva de agua y electrolitos (10).
5. Enfermedades asociadas a la alteración del transporte intestinal
Las alteraciones en el transporte intestinal de agua y electrolitos son un componente fisiopatológico a destacar en diversas enfermedades gastrointestinales. Estas alteraciones pueden deberse a mecanismos infecciosos, inflamatorios, genéticos o estructurales, y afectan tanto la absorción como la secreción, generando desequilibrios hídricos que se manifiestan clínicamente en forma de diarreas agudas o crónicas, malabsorción o deshidratación severa; o complicaciones más graves que generan diversas afecciones, como la enfermedad de Crohn o el Síndrome del Intestino Corto (1).
5.1. Enfermedad de Crohn
La enfermedad de Crohn (EC) es una afección inflamatoria crónica del intestino identificada inicialmente como ileítis regional por Crohn, Ginzburg y Oppenheimer a principios del siglo pasado. La inflamación de la EC compromete cualquier segmento del tracto gastrointestinal, siendo el íleon distal la zona frecuentemente afectada. Se caracteriza por la alternancia de períodos de brotes y de remisión durante el curso de su enfermedad. Su patogénesis resulta de las interacciones de factores ambientales, sistema inmunitario, genes de susceptibilidad y cambios en el microbioma del huésped, lo que lleva a la alteración de la mucosa intestinal (12, 13).
La evolución de la EC implica una alteración compleja de la respuesta inflamatoria, con disfunción de la inmunidad innata de la barrera mucosa intestinal y remodelación de la matriz extracelular mediante metaloproteínas y moléculas de adhesión como MAdCAM-1. Este entorno favorece la migración leucocitaria y una respuesta Th1, mediada por citocinas como IL-12 y TNF-α (12, 13).
La EC se origina a partir de una inflamación tisular desencadenada por una respuesta inmunitaria desregulada frente a antígenos bacterianos presentes en la luz intestinal. (Figura 2). Diversas células inmunitarias (T CD4 y CD8, células B, monocitos CD14 y células asesinas naturales) participan en este proceso al infiltrarse en el tejido intestinal. Una parte de la susceptibilidad inmunomediada en la EC se relaciona con mecanismos de defensa innata frente a infecciones, entre los cuales destaca la secreción de moco intestinal. En modelos murinos, se ha evidenciado que las variantes del gen Muc2 que disminuyen la producción de moco se asocian con el desarrollo de la EC (12,13,15).
Asimismo, se ha correlacionado con las moléculas implicadas en la adhesión bacteriana. Un ejemplo de ello es el gen FUT2, que codifica la enzima fucosiltransferasa, encargada de la secreción de formas solubles de los antígenos del sistema ABO. Las personas con variantes del gen FUT2 que reducen la secreción de antígenos presentan una interacción bacteriana alterada, lo que incrementa su predisposición a desarrollar EC (12, 13).
Además, la EC también involucra la interacción de estas células con integrinas, moléculas de adhesión y diversas quimiocinas, que inducen una producción elevada de citocinas inflamatorias, lo cual activa células inmunes y no inmunes, así como la inflamación de la mucosa. Entre las diversas moléculas de adhesión, existen evidencias que sugieren la participación del leucocito MAdCAM-1, receptor de la integrina α4β4, en la patogenia de la enfermedad (12, 13).
Por otro lado, la matriz extracelular también interviene en la activación leucocitaria, mediante proteínas como CD44, CD26 y metaloproteinasas, especialmente MMP1 y MMP3, abundantes en zonas inflamadas. (12, 15).
En la mucosa de pacientes con EC se detecta una desregulación constante del sistema inmunitario, destacando una marcada hiperactividad de los linfocitos T y una producción excesiva de citocinas, como IL-12 e IFN-γ, que promueven un fenotipo TH1. También se ha demostrado que el TNF-α incrementa la cantidad de linfocitos T reg CD4+ FoxP3+, especialmente en la mucosa intestinal de niños con la EC. La inhibición de TNF-α reduce los efectos dañinos en ciertos subgrupos de pacientes con EC. Asimismo, en pacientes con EC, se observa expresión anormal de interleucinas, que regulan el crecimiento, la maduración y la activación de distintas poblaciones celulares (12, 13, 15).
Análisis adicionales de subgrupos de células T han revelado la presencia de células TH 1 y TH 17 en la EC, siendo TNF, IL-12 e IL-23, las citocinas más implicadas. También se ha asociado la IL-34, cuya expresión es más pronunciada en zonas de inflamación activa, donde induce la expresión de TNF-α e IL-6 a través de una vía mediada por ERK; además, induce la producción de CCL20 mediante su interacción con 3l receptor M-CSFR1, expresado en el epitelio colónico inflamado, pero ausente en controles sanos (12, 13, 15).
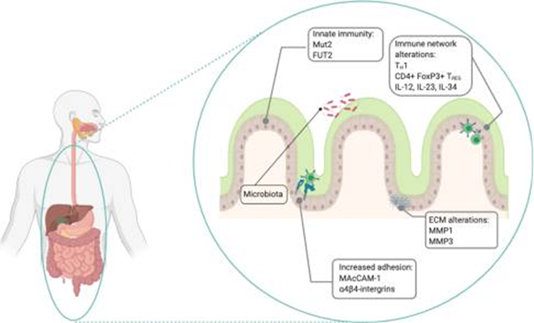
Figura 2. Patogénesis de la enfermedad de Crohn. La inmunidad innata está implicada en alteraciones de la barrera mucosa intestinal, vinculadas a genes como Muc2 y FUT2, mientras que la inmunidad adaptativa se caracteriza por una respuesta mediada por linfocitos TH1 y células T reg, influida por citocinas como TNF-α, IL-12, IL-34 e IL-23. La migración celular hacia los sitios inflamatorios se ve favorecida por la remodelación de la matriz extracelular, a través de la acción de metaloproteinasas como MMP-1 y MMP-3, y por la sobreexpresión de moléculas de adhesión, entre ellas MAdCAM-1 e integrina α4β4. Por último, la interacción entre el epitelio intestinal y la microbiota se ha asociado con la progresión de la enfermedad.
Los linfocitos T son los principales efectores en la inflamación del tejido intestinal; sin embargo, el sistema inmunitario humoral también participa en este proceso. En particular, los linfocitos T CD4+ inducen la diferenciación de células plasmáticas mediante un mecanismo altamente dependiente de la IL-2, una citocina que se encuentra aumentada en el intestino de pacientes con EC. La IL-21 induce la diferenciación de linfocitos B vírgenes en linfocitos B productores de granzima B, la cual posee una actividad citotóxica en la mucosa intestinal y perpetúa el daño epitelial (12, 13, 15).
5.2. Diarrea secretora
Las enfermedades diarreicas siguen siendo un importante problema de salud pública mundial, especialmente en niños menores de 5 años y adultos mayores de 70, en quienes la mortalidad se ve agravada por factores como la desnutrición y las infecciones entéricas previas. Su prevalencia está estrechamente relacionada con el clima y el desarrollo económico, donde predominan causas infecciosas como rotavirus; principal agente de diarrea grave a nivel mundial, Vibrio cholerae, Escherichia coli enterotoxigénica, Shigella, Salmonella y parásitos como Entamoeba histolytica (16). En el Perú, hasta la SE 03-2024 se notificaron 70 837 episodios y 10 muertes por EDA, cifras similares al mismo periodo de 2023, cuando se registraron 71 028 casos y 5 muertes (17).
La diarrea se produce por un desequilibrio entre secreción y absorción de líquidos y electrolitos a través del epitelio intestinal, proceso regulado por el transporte activo de iones como Na⁺, Cl⁻, HCO₃⁻ y K⁺, así como de solutos como la glucosa. Las diarreas secretoras pueden tener origen bacteriano, viral o congénito, con mecanismos fisiopatológicos distintos pero que convergen en la alteración del transporte intestinal de agua y electrolitos (18).
En primer lugar, mencionamos a las diarreas bacterianas, en las que bacterias como Vibrio cholerae y Escherichia coli enterotoxigénica producen enterotoxinas específicas, como la toxina del cólera y la enterotoxina termoestable, que elevan los niveles intracelulares de nucleótidos cíclicos, activando los canales apicales CFTR de Cl⁻ y estimulando su secreción. Estudios en células intestinales humanas, demuestran que el aumento de AMPc, GMPc y Ca²⁺ inducido por estas toxinas también inhibe el intercambiador NHE3. Además, algunas bacterias incrementan la liberación de agonistas humorales, neurotransmisores o neuropéptidos, como la 5-hidroxitriptamina, los péptidos VIP y el receptor de galanina tipo 1, potenciando la secreción de Cl⁻ y reduciendo la absorción de Na⁺ (Figura 3a). Por su parte, bacterias invasoras como Salmonella y Shigella generan inflamación tisular mediante el reclutamiento de células inmunes y la liberación de citocinas, lo que activa vías de señalización intracelular dependientes de Ca²⁺ (19).
En segundo lugar, presentamos a las diarreas virales, la cual es causada principalmente por el rotavirus entérico, que provoca tanto secreción excesiva de fluidos como alteraciones estructurales en el epitelio intestinal, originando una diarrea secretora con marcada influencia de la edad (20). Estudios han propuesto que una proteína viral, NSP4, actúa como enterotoxina al incrementar la concentración citoplasmática de Ca²⁺ mediante su unión a receptores de membrana como la integrina α1β2, la interacción con el neuropéptido galanina y/o la activación de nervios entéricos (Figura 3b). Además, NSP4 inhibe los transportadores NHE3 y SLC5A1, afectando la absorción de sodio y glucosa (19).
Finalmente, tenemos a las diarreas congénitas; estas pueden originarse por mutaciones genéticas hereditarias poco frecuentes que afectan diversas proteínas intestinales (Figura 3c). El avance de la secuenciación genética de bajo costo ha permitido identificar los genes implicados en muchas de estas enteropatías familiares. Por ejemplo, mutaciones en el transportador de sodio SLC5A1 provocan malabsorción de glucosa y galactosa, mientras que alteraciones en el intercambiador de cloruro/bicarbonato DRA generan diarrea congénita por pérdida de Cl⁻. Asimismo, mutaciones en el receptor de guanilina GUCY2C, responsables del síndrome de diarrea familiar, inducen una activación constitutiva de cGMP que estimula la secreción de Cl⁻ mediada por CFTR e inhibe el transportador NHE3 (19).
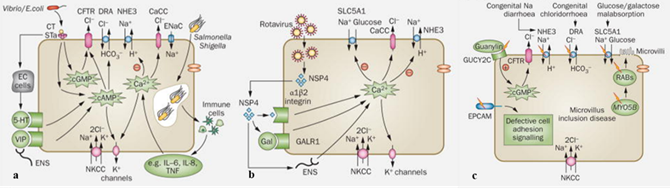
Figura 3. Mecanismos de absorción y secreción de fluidos intestinales en diarreas secretoras. (a) Diarrea bacteriana. Algunas bacterias liberan enterotoxinas que elevan los niveles de nucleótidos cíclicos intracelulares, lo que favorece la secreción de Cl⁻ e inhibe la absorción de Na⁺ mediada por NHE3. Por su parte, las bacterias invasoras desencadenan una respuesta inflamatoria tisular que involucra el reclutamiento de células inmunes y la liberación de citocinas, activando así vías de señalización intracelular dependientes de Ca²⁺. (b) Diarrea causada por rotavirus entérico. La proteína NSP4 del rotavirus eleva la concentración citoplasmática de Ca²⁺ al unirse a la integrina α1β2, interactuar con la galanina y/o activar los nervios entéricos. Además, inhibe los transportadores NHE3 y SLC5A1. (c) Diarrea Congénita. Las diarreas congénitas se originan por raras mutaciones genéticas hereditarias que afectan diversas proteínas intestinales.
5.3. Síndrome del intestino corto
En el Síndrome de Intestino Corto (SIC) se producen la incapacidad para absorber adecuadamente macronutrientes y micronutrientes, conlleva deficiencias vitamínicas y minerales, desequilibrios hidroelectrolíticos y pérdida de hormonas gastrointestinales que regulan la motilidad, el tránsito intestinal y el vaciamiento gastroduodenal; la severidad del compromiso funcional depende de la longitud del intestino remanente, los segmentos conservados, la calidad de absorción y la capacidad adaptativa individual (21).
Cuando existe colon en continuidad, este adquiere un papel digestivo esencial al reabsorber agua, electrolitos y ácidos grasos de cadena corta (SCFA) provenientes de la fermentación bacteriana de carbohidratos no absorbidos; hasta el 65 % de la ingesta de hidratos puede perderse si no se fermenta, aportando hasta 500 kcal/día y, mediante hormonas como PYY y GLP-2, frenando el tránsito y estimulando la adaptación; en cambio, en pacientes con yeyunostomía sin colon, cualquier remanente yeyunal inferior a 100 cm puede convertirse en “secretor neto” con alta pérdida de líquidos (21). La pérdida de la válvula ileocecal facilita el sobrecrecimiento bacteriano en intestino delgado por dilatación y enlentecimiento, con consecuencias como competencia por nutrientes, inflamación, hemorragia, translocación bacteriana, endotoxemia, daño hepático y acidosis D-láctica. Tras la resección yeyunal, se produce una reducción transitoria de la absorción hasta que el intestino restante se adapta, pero soluciones hiperosmolares exacerban las pérdidas de líquidos y la pérdida de hormonas de retroalimentación gástrica acelera el vaciamiento de líquidos, sobrepasando la capacidad absortiva remanente. Mientras que la resección ileal compromete la reabsorción de las 7 L de secreciones diarias, provoca una deficiencia de la vitamina B12 y perdida de las sales biliares, de modo que, con menos de 100 cm de íleon, se produce una respuesta secretora neta al alimento, malabsorción de grasas y pérdida de los frenos hormonales ileocólicos (PYY, GLP-1, neurotensina), acelerando el tránsito y agravando la diarrea (22).
El síndrome de intestino corto puede tener múltiples etiologías: resecciones quirúrgicas extensas en enfermedad de Crohn o tras eventos de isquemia mesentérica masiva; trastornos malabsortivos sin acortamiento anatómico como la pseudo-obstrucción intestinal crónica, el sprue refractario, la enteritis por radiación o la atrofia vellosa congénita; también se mencionan traumatismos abdominales, neoplasias, radioterapia o íleo obstructivo. En el adulto sano, el intestino delgado mide entre 275 y 850 cm y absorbe la mayor parte de nutrientes y aproximadamente 7 L de líquidos diarios; se considera que existe SIC cuando el remanente es inferior a 180–200 cm, situación que conduce a insuficiencia intestinal y deficiencias nutricionales (23). La insuficiencia intestinal se define como la incapacidad de absorber suficiente energía pese a un aumento en la ingesta, o la imposibilidad de adaptarla a la función reducida del intestino, requiriendo soporte intravenoso para cubrir las necesidades. Clínicamente, se clasifica en tipo 1 (aguda y autolimitada), tipo 2 (aguda prolongada, con nutrición parenteral necesaria por semanas o meses) y tipo 3 (crónica, reversible o irreversible, con dependencia de nutrición parenteral a largo plazo) (23).
5.4. Adaptación del intestino delgado.
Tras la resección intestinal en el síndrome de intestino corto, la malabsorción inicial mejora gracias a cambios adaptativos en la mucosa remanente, que comienzan tan pronto como a las 48 horas posoperatorias. En la adaptación estructural, se observa hipertrofia e hiperplasia de las vellosidades para expandir el área de absorción, así como dilatación progresiva del lumen; pero la apoptosis de los enterocitos no disminuye, aunque puede moderarse mediante péptidos como el factor de crecimiento epidérmico (EGF). Paralelamente, la adaptación funcional involucra el incremento de sistemas de transporte de nutrientes. Por ejemplo, un aumento hasta dos veces en la cotransportación sodio–glucosa (SGLT-1), la aparición de nuevos transportadores dependientes de sodio tanto para glucosa en el íleon como para oligopéptidos (PepT1) en el colon, la elevación de intercambiadores electroneutrales de sodio/hidrógeno (NHE-3) y de mensajeros de acuaporinas (Figura 1). Este proceso puede prolongarse hasta dos años para que el paciente supere la insuficiencia intestinal y reduzca su dependencia de la nutrición parenteral (22). Dado que la presencia de nutrientes en la luz intestinal es un estímulo necesario, la nutrición enteral temprana favorece la proliferación de células de la criptas mediada por factores tróficos liberados en respuesta a alimentos y secreciones biliares y pancreáticas. Entre los nutrientes tróficos se incluyen glutamina, un sustrato primordial para la síntesis de ácidos nucleicos y activador de MAP quinasas; ácidos grasos de cadena corta, ácidos grasos insaturados, ornitina y nucleótidos. Además, el rol de factores de crecimiento sistémicos y locales es crítico: hormonas enterales como enteroglucagón, colecistoquinina, gastrina, EGF, Hb-EGF, factor de crecimiento de queratinocitos, neurotensina, leptina, hormona de crecimiento, IGF-1 y -2, PYY, GLP-2 e insulina contribuyen a orquestar la respuesta adaptativa; en particular, el péptido GLP-2, ya que eleva el flujo sanguíneo intestinal y revierte la atrofia vellosa, potenciando el índice vellosidad/criptas y favoreciendo la ganancia de peso y la absorción proteica (24).
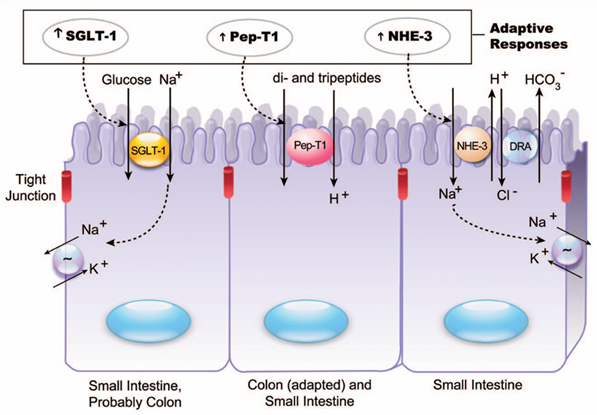
Figura 4. Cambios adaptativos en los mecanismos de transporte celular epitelial. Se observa un aumento de la captación de glucosa acompañada de sodio mediante SGLT-1 en el intestino delgado (y posiblemente en el colon), así como una mayor absorción de di- y tripéptidos tanto en el intestino delgado como en el grueso. Además, mejora la reabsorción electroneutral de NaCl en el intestino delgado gracias a la mayor actividad de los intercambiadores sodio/hidrógeno (NHE-3) y cloruro/bicarbonato (DRA).
CONCLUSIONES
· El transporte intestinal de fluidos y electrolitos constituye un proceso esencial para la homeostasis del organismo, sustentado en mecanismos altamente regulados que permiten una absorción eficiente de agua y solutos, así como una secreción controlada en función de las necesidades fisiológicas. Esta función se ejecuta gracias a la acción coordinada de transportadores como SGLT1, NHE3, DRA, ENaC y canales como CFTR, regulados por señales neurohormonales y paracrinas.
· Se ha evidenciado que el intestino delgado participa mayormente en la absorción isoosmótica de sodio, cloruro y agua, mientras que el colon cumple un rol decisivo en la reabsorción fina de electrolitos y la conservación de volumen corporal, sobre todo en estados de déficit hidrosalino. Esta dinámica depende de mecanismos celulares precisos, como el cotransporte sodio-glucosa, los intercambiadores Na⁺/H⁺ y Cl⁻/HCO₃⁻, y la actividad hormonal de sustancias como la aldosterona y el péptido natriurético.
· Asimismo, la alteración de estos mecanismos da lugar a importantes patologías clínicas. En la diarrea secretora, se activan vías intracelulares que estimulan la secreción masiva de cloro y agua a través de CFTR, mientras que en la enfermedad de Crohn, la inflamación crónica inhibe transportadores absortivos y compromete la integridad de la barrera epitelial. Por otro lado, el síndrome de intestino corto demuestra cómo la pérdida anatómica intestinal obliga a una adaptación estructural y funcional del intestino remanente, donde el conocimiento del transporte de solutos es clave para el manejo nutricional y terapéutico.
· Finalmente, comprender los fundamentos fisiológicos del transporte intestinal y su implicancia en patologías permite no solo el diagnóstico y tratamiento más efectivo, sino también la aplicación de estrategias preventivas y terapias dirigidas, como la rehidratación oral, la nutrición enteral precoz o el uso de moduladores hormonales, que resultan fundamentales en la práctica clínica moderna.
REFERENCIAS BIBLIOGRÁFICAS
1. Guyton AC, Hall JE. Tratado de fisiología médica. 13.ª ed. Barcelona: Elsevier; 2017.
2. Boron WF, Boulpaep EL. Fisiología médica. 3.ª ed. Elsevier España; 2017.
3. Intestinal Ion and Nutrient Transport in Health and Infectious Diarrhoeal Diseases. J Physiol. 2012;590(10):2189-2200. https://pubmed.ncbi.nlm.nih.gov/3069442/
4. Barrett KE, Barman SM, Brooks HL, Yuan JX. Fisiología médica de Ganong. 26.ª ed. México: McGraw-Hill Education; 2020.
5. Kiela PR, Ghishan FK. Physiology of Intestinal Absorption and Secretion. Best Pract Res Clin Gastroenterol. 2016;30(2):145-159. https://pmc.ncbi.nlm.nih.gov/articles/PMC4956471/
6. McLaughlin J. Gastrointestinal physiology. Surgery (Oxford). 2009;27(6):225-230. https://pmc.ncbi.nlm.nih.gov/articles/PMC4956471/
7. Physiology of the Gastrointestinal System. In: Comprehensive Toxicology. 3rd ed. Elsevier; 2018. p. 17–41. https://pure.hud.ac.uk/en/publications/physiology-of-the-gastrointestinal-system
8. Cavin JB, Cuddihey H, MacNaughton WK, Sharkey KA. Acute regulation of intestinal ion transport and permeability in response to luminal nutrients: the role of the enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2020;318(2):G254–64. https://journals.physiology.org/doi/full/10.1152/ajpgi.00186.2019?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
9. Roberts ML, et al. Cortisol’s role in intestinal electrolyte transport and stress adaptation. J Endocrinol. 2019;243(1): R1–12.
10. Fung C, Vanden Berghe P. Functional circuits and signal processing in the enteric nervous system. Cell Mol Life Sci. 2020;77(22):4505–22. Disponible en: https://link.springer.com/article/10.1007/s00018-020-03543-6
11. Nakai D, Miyake M. Intestinal membrane function in inflammatory bowel disease. Pharmaceutics. 2023;15(11):2597. https://www.mdpi.com/1999-4923/16/1/29
12. Pasternak G, Chrzanowski G, Aebisher D, Myśliwiec A, Dynarowicz K, Bartusik-Aebisher D, et al. Crohn’s disease: basic characteristics of the disease, diagnostic methods, the role of biomarkers, and analysis of metalloproteinases: a review. Life. 2023;13(10):2062. https://www.mdpi.com/2075-1729/13/10/2062
13. Petagna L, Antonelli A, Ganini C, Bellato V, Campanelli M, Divizia A, et al. Pathophysiology of Crohn’s disease inflammation and recurrence. Biol Direct. 2020;15:23. https://pmc.ncbi.nlm.nih.gov/articles/PMC7648997/
14. Sánchez Bonilla E, Wong Álvarez ÓF, Fung Cai M. Enfermedad de Crohn: un enfoque integral en su patogenia, diagnóstico y tratamiento. Rev Méd Sinergia. 2023 ago [citado el 6 ago 2025];8(8):e1092. Disponible en: https://revistamedicasinergia.com/index.php/rms/article/download/1092/2272/7835
15. Silva F, Gatica T, Pavez C. Etiología y fisiopatología de la enfermedad inflamatoria intestinal. Rev Med Clin Condes. 2019;30(4):262–72. https://www.elsevier.es/es-revista-revista-medica-clinica-las-condes-202-articulo-etiologia-y-fisiopatologia-de-la-S0716864019300574
16. Riverón Corteguera RL. Fisiopatología de la diarrea aguda. Revista Cubana Pediátrica. 1999;71(2):86‑115. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75311999000200005
17. Centro Nacional de Epidemiología, Prevención y Control de Enfermedades. Enfermedad diarreica aguda, Perú: Boletín epidemiológico, SE 03-2024. Lima: Ministerio de Salud del Perú; 2024. Disponible en: https://www.dge.gob.pe/portal/docs/vigilancia/sala/2024/SE03/edas.pdf
18. Thiagarajah JR, Donowitz M, Verkman AS. Secretory diarrhoea: mechanisms and emerging therapies. Nat Rev Gastroenterol Hepatol. 2015;12(8):446‑57. Disponible en: https://pmc.ncbi.nlm.nih.gov/articles/PMC4786374/
19. Keely SJ, Barrett KE. Intestinal secretory mechanisms and diarrhea. Am J Physiol Gastrointest Liver Physiol. 2022;322(4): G405‑20. Disponible en: https://journals.physiology.org/doi/full/10.1152/ajpgi.00316.2021
20. Whyte LA, Jenkins HR. Pathophysiology of diarrhoea. Paediatr Child Health (Oxford). 2012;22(10):443‑7. Disponible en: https://www.sciencedirect.com/science/article/abs/pii/S175172221200087X
21. Donohoe CL, Reynolds JV. Short bowel syndrome. The Surgeon. 2010;8(5):270–279. doi:10.1016/j.surge.2010.06.004
22. Navarro F, Gleason WA, Rhoads JM, Quiros-Tejeira RE. Short bowel syndrome: Epidemiology, pathophysiology, and adaptation. Neoreviews 2009;10;e330. DOI: 10.1542/neo.10-7-e330
23. Lakkasani S, Seth D, Khokhar I, Touza M, Dacosta TJ. Concise review on short bowel syndrome: Etiology, pathophysiology, and management. World Journal of Clinical Cases. 2022;10(31):11273–11282. doi:10.12998/wjcc.v10.i31.11273
24. Tappenden KA. Pathophysiology of short bowel syndrome. Journal of Parenteral and Enteral Nutrition. 2014;38(1 Suppl):14S–22S. doi:10.1177/0148607113520005